BioHub
| Author: | Diane Trout |
|---|
This presentation and associated material is available at http://woldlab.caltech.edu/biohub/scipy2006/
Motivation: Chr 1
>chr1 taaccctaaccctaaccctaaccctaaccctaaccctaaccctaacccta accctaaccctaaccctaaccctaaccctaaccctaaccctaaccctaac cctaacccaaccctaaccctaaccctaaccctaaccctaaccctaacccc taaccctaaccctaaccctaaccctaacctaaccctaaccctaaccctaa ccctaaccctaaccctaaccctaaccctaacccctaaccctaaccctaaa ccctaaaccctaaccctaaccctaaccctaaccctaaccccaaccccaac cccaaccccaaccccaaccccaaccctaacccctaaccctaaccctaacc ctaccctaaccctaaccctaaccctaaccctaaccctaacccctaacccc taaccctaaccctaaccctaaccctaaccctaaccctaacccctaaccct aaccctaaccctaaccctcgcggtaccctcagccggcccgcccgcccggg
Motivation: Rest of human
That was the first 500 DNA bases of human chromosome 1, and there are about 3.2 billion more just like those.
And that's for one species.
- Current estimates put humans at about 25,000 genes
Motivation: Genome sizes
Order of genome size in millions of bases (Mb).
| Class | Genome size |
|---|---|
| Wheat | 15000 |
| Mammals | 1000 |
| Worm, Fruit fly | 100 |
| Yeast | 10 |
| Bacteria | 1 |
|
0.580 |
- Genitalium has about 470 genes
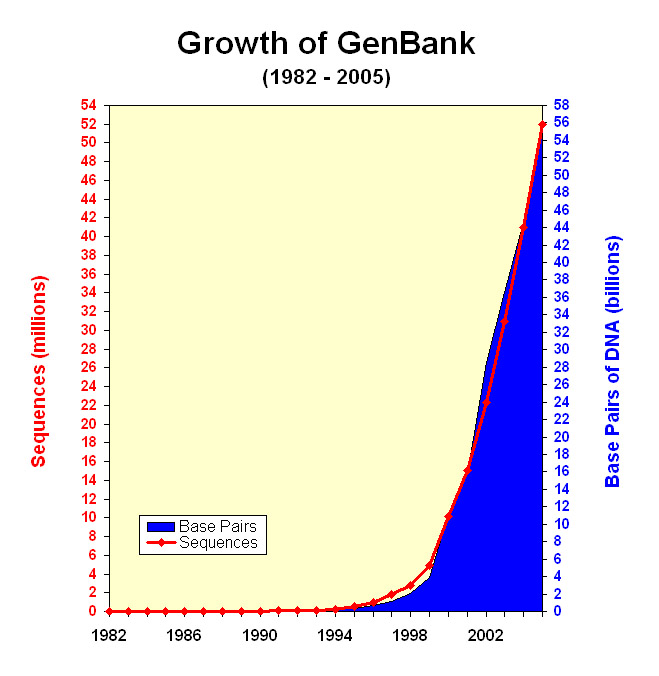
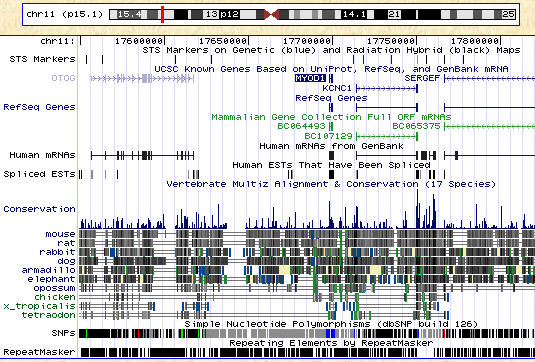
Motivation: Genome Browser
Introducing: BioHub
The purpose of BioHub is to
- provide a python interface for large-scale genomic analyses
- provide a database to link diverse annotation sources to specific genome locations.
- (AKA local or remote databases)
Core Concepts
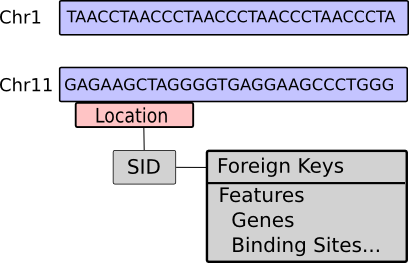
- A BioHub Genome is a collection of fasta files storing each chromosome.
- A genome is named by species and version number.
- A location is a genome + chromosome + start + stop + strand
- We always return the same sequence ID (SID) for the same location.
Since there are two strands to a DNA molecule but one strand completely determines the other, when storing sequence we need only store one strand.
However when storing a location we need to indicate if we are stored on the "forward" or "reverse" strand.
BioHub Genome
Human 34.0
Example
To illustrate how BioHub can be used for a large scale genomic analysis we'll construct a simple example.
- Create an annotation database
- Link the annotations via BioHub
- Locate sequence and other annotations "near" our annotations.
Create Sample Database
Imagine we have the results from some microRNA scanning programs:
class QueryFasta:
def __init__(self, query_fasta):
class miRNATarget:
def __init__(self, mirna_name, binding,
query_seq, target_seq):
microRNAs are one regulator of gene expression.
Create Sample Database
Start with a simple "database":
# sequence fed to target finder
gene2995 = QueryFasta(">GeneID:2995\nCAC...")
results2995 = []
results2995.append(
miRNAResult("hsa-mir-140", #name
-21.2, # strength
gene2995, # seq searched
"CAAGA..." # seq found
))
# sequence fed to microRNA target finder
gene2995 = QueryFasta(""">GeneID:2995
CACTGCATTTCCCTTTACCAACTAGCGCTGGGAGCACTGGACACTTAAA
TCCTCATCTGTCCTCCTTTCCTGTAAATAAAAGCCCTTCTATCCA""")
results2995=[]
results2995.append(
# result, name, binding strength,
# query sequence, and miRNA target
miRNAResult("hsa-mir-140", # result name
-21.2, # binding strength
gene2995, # sequence we searched
# sequence we found
"CAAGATATTACCATGTACATGGTACCACCATC"
))
# store another sequence
results2995.append(
miRNAResult("hsa-mir-206", -20.4, gene2995,
"TCTTACCCATGAATGTGCACTACCTACATTTT"))
cPickle.dump(results2995,
"gene2995_mirna.pickle")
Registration
- Registration is how we tell BioHub to generate an ID for a particular
chunk of genome.
- registerSequenceGivenLocation
- registerSequenceByBlast
Registration
registerSequenceGivenLocation(self, locationList, # (start,stop) description, # desc. of run user, # username sequenceType, # e.g. CDS, Intron contig=None, # Contig object species=None, # Name of species build=None, # Genome version accession=None, # NCBI accession gi=None, # NCBI gi id=None, # chromosome names reverseComplement=False)
You don't need specify all of these at the same time.
Registration
Register by BLAST
registerSequenceByBlast(self, sequenceString, # search sequence species, # Name of species build, # Genome version seqType, # Desc of seq desc, # Desc of Registration user, # username header=None) # optional fasta header
Throws an exception when more than one location is found.
BLAST
BLAST is a sequence (string) search tool that allows for mismatches, insertions and deletions.
Because of this inexact matching in larger genomes you need 20-40 bases to avoid purely random matches.
Link Annotations
- One problem with microRNAs is that they are typically the length where one would start to expect matches by random chance.
- Thus we should search for the location of our longer "query" sequence first, and then find the offset for our miRNA target
this code will attempt to find the exact genomic location for our miRNAs:
from BioHub.Util.BlastDB import BlastDB
from BioHub.Util.BlastHandler import NMismatchesBlastHandler
blast_filter = NMismatchesBlastHandler(0)
# create a reference to a blast database
human_db = BlastDB("Homo sapiens build 34")
# go run blast
blast_handle = human_db.blastBySequence(gene2995.sequence)
# parse the miserable BioPython blast handler results
matches = blast_filter.getMatchesGivenBlastFileHandle(blast_handle)
if len(matches) != 1:
raise SequenceNotFound("too many copies of query sequence found")
# convert high scoring pair to location
# FIXME: this example got to complicated and this hasn't been
# FIXME: implemented yet. Check for updates on our website.
contig, start, stop, strand = hspToLocation(matches[0][1])
seq2995 = gene2995.sequence
for mirna_result in results2995:
target_seq = mirna_result.target_seq
reverseCompliment = false
target_start = seq2995.find(target_seq)
# figure out which strand we're on
if target_start == -1:
target_reverse_seq = reverse_compliment(target_seq)
target_start = seq2995.find(target_reverse_seq)
reverseCompliment = true
if target_start == -1:
raise SequenceNotFound("I give up")
target_start = start+target_start
target_stop = start+target_start+len(target_seq)
sid = genome.RegisterSequenceByLocation()
biohub_link[mirna_result] = sid
- but for simplicity we'll just pretend there is enough specificity
Link Annotations
registrar = RegisterSequence()
for mirna_result in results2995:
target_seq = mirna_result.target_seq
sid = registrar.registerSequenceByBlast(
target_seq,
"Human",
34,
"miRNA",
"found a microRNA",
"diane")
biohub_link[mirna_result] = sid
from BioHub.BioHubAPI.RegisterSequence \
import RegisterSequence
registrar = RegisterSequence()
for mirna_result in results2995:
target_seq = mirna_result.target_seq
sid = registrar.registerSequenceByBlast(
target_seq,
"Human",
34,
"miRNA",
"found a microRNA",
"diane")
biohub_link[mirna_result] = sid
Perform Query
a microRNA always targets part of a gene, so lets check to make sure we actually are contained in a gene.
sidList = SpatialQuery.getSidsContainingSid(
registered_sid,
seqTypes=['gene'],
strand=SpatialQuery.STRAND_BOTH)
assert( len(sidList) != 0 )
# Find what gene we're contained in
from BioHub.BioHubAPI import SpatialQuery
for result, registered_sid in biohub_link.items():
sidList = SpatialQuery.getSidsContainingSid(
registered_sid,
seqTypes=['gene'],
strand=SpatialQuery.STRAND_BOTH)
assert( len(sidList) != 0 )
Perhaps we might want to find the next upstream gene:
nextGeneSid = SpatialQuery.getSidNextTOSid(
result_sid,
searchDirection = SpatialQuery.DIRECTION_UPSTREAM,
seqTypes=['gene'],
maxBP=10000,
strand=SpatialQuery.STRAND_SAME)
Or we might want to find all annotations 10Kbps downstream:
downstreamSIDs = SpatialQuery.getSidsNextToSid(
result_sid,
downstreamBp=10000)
Perform Query
Search for Motifs (regulatory elements) near our microRNA.
regSIDs = getSidsNextToSid(
result_sid,
upstreamBp=10000, downstreamBp=10000,
seqTypes=['motif', 'binding_site',
'conserved'],
strand=STRAND_BOTH,
inclusive=True, # include us
# only include fully in
overlap=OVERLAP_EXACTLY_IN)
Advanced BioHub Workflow
- Search genome for genes with specific regulatory binding site (like a microRNA)
- Find a set of genes that interact
- Extract likely microRNA target region
- Feed likely targets to multiple microRNA prediction tools.
- Anything look promising?
- Put down the computer and perform wet bench experiments
- Record results in BioHub
- Next lab user begins a search with validated microRNA targets...
Future Work
- More unit tests
- Streamline API
- Consider using SQLAlchemy instead of our own ORM
- Support migrating annotations between genome builds
- Homology database
- Gene ontology database
- Link to expression database
- BioHub Trac - http://woldlab.caltech.edu/cgi-bin/biohub/ticket/
Acknowledgments
| Core Development | PI |
|---|---|
|
|
|
|
|
|
|
|
|
Project Page: http://woldlab.caltech.edu/biohub/
Support
- Department of Energy
- NIH GMS
- NASA
- Moore Minority Scholars Program